|
NSF Proposal - 7. Phylogenetic Analysis
Principles of OTU and character selection.
Due to the integrative nature of the proposed analyses, in which
data from many sources will be considered, the concepts of "OTU"
and character will vary within and among datasets. Data at this
level are always compiled from study of different organisms considered
to represent the same OTU. Thus OTUs are always composites in practice;
their composition varying depending on the scale of analysis. Likewise,
what counts as a useful character changes depending on the scale
of analysis. The columns in a data matrix are already refined hypotheses
of phylogenetic homology. There is also a clear reciprocal relationship
between OTUs and characters. An OTU can best be defined as a set
of individual samples that are homogeneous for characters currently
known, while a character can be defined as a potential marker for
shared history of some subset of the known OTUs. This means that
OTUs and characters emerge during a process of "reciprocal
illumination." To a large extent their definitions are interlinked,
so how do we proceed empirically in a way that avoids circularity"
We will take great care to examine both concepts of character and
OTU in the proposed research. We will use the relatively advanced
state of knowledge of characters and phylogenetic structure in the
green plants as a model system for testing alternative approaches
to analysis in a systematic manner. The overarching goal is to develop
ways to scale OTU composition and character definition up and down
the many fractally-nested levels making up the tree of life.
Character analysis. Phylogenetic
analysis can be broken down into two discrete phases: character
analysis and cladistic analysis. In the former phase, a data matrix
is assembled as discussed above. Potential characters are evaluated
by rules of character analysis, an evaluation of evidence for:(1)
homology and heritability of a character across the taxa being studied,
(2) independent evolution of different characters, and (3) presence
in each character of a system of at least two discrete states. These
criteria will be applied here to data matrices assembled at several
scales of analysis. The deepest scale will be a matrix of the ca.
50 exemplar green plants plus outgroups (see section 4), with much
of the data newly generated from this proposal. These OTUs will
be thoroughly studied for the morphological characters covered above,
and have completely sequenced mitochondrial and chloroplast genomes.
We will also have nuclear BAC libraries constructed for most exemplars,
which will facilitate discovering gene translocations between the
organellar genome and the nuclear genome, and sequencing of new
candidate nuclear genes. Characters will be evaluated in the following
categories:
- Genomic characters. Structural genomic differences resulting
from inversions, translocations, gene losses, duplications, and
insertion/deletion of introns will be identified within and between
the three genomes and likely homologies established (e.g, examining
the ends of breakpoints to see whether a single event is likely
to have occurred).
- Morphological characters. All features that can be compared
across this deep level of analysis will be evaluated for independence
and discrete states. The literature will be used, but wherever
possible original material will be reexamined.
- DNA sequence data. To compare DNA sequence characters with
genomic and morphological characters, we will also align all genes
available in the three genomes, We will do this two ways (in order
to compare results): a liberal alignment using as much sequence
as possible, and a conservative alignment using only regions that
are unambiguously alignable. Both amino acid and nucleotide alignments
will be analyzed where appropriate for protein coding genes.
Matrices will be developed for local clades using data appropriate
at that level. These data will come almost entirely from other research
groups and collaborators as discussed in the management plan section.
Cladistic analysis. The second
phase of phylogenetic analysis involves turning data matrices into
a recontructions of a phylogenetic tree. We will explore the full
spectrum of approaches to building phylogenetic trees from data
matrices and how to concatenate the results from the different scales
of phylogenetic analysis to be undertaken here. We will use only
character-based methods of phylogenetic analysis, and mainly work
within a maximum parsimony framework (given the very heterogeneous
set of characters). However, we will compare and contrast equal
and differentially-weighted parsimony and maximum likelihood methods
as applied to DNA sequence data. The first task will be to analyse
the data matrix of the ca. 50 exemplar taxa, and the mixture of
genomic, morphological, and DNA sequence characters discussed above,
to produce a "backbone" phylogeny of basal green plants.
The next task will be to use this sparsely-sampled, but extremely
character-rich, global phylogeny to connect up all the many local
phylogenetic data sets available from other research groups. These
local data sets sample many more taxa (thousands taken all together),
but with considerably less character data available.
For this second task we will assemble all published phylogenetic
trees on the relevant chlorophyte and streptophyte lineages (e.g.,
references above), and will closely coordinate our efforts with
ongoing phylogenetic projects of direct relevance to ours (see "Management
Plan"). We will insure that all relevant phylogenetic studies
are entered into TreeBASE (www.treebase.org), thereby providing
ready access to project members (and the entire scientific community)
to phylogenetic knowledge on green plant lineages.
Concatenation analyses. This
assembly of individual phylogenetic trees and data sets will be
critical to the construction of large-scale concatenated trees.
In collaboration with M. Sanderson (UC Davis) we will use green
plant phylogenies to explore a variety of algorithms for producing
supermatrices and supertrees, such as Matrix Representation Parsimony
(e.g., [133-135]) and methods that can take branch lengths into
account [136]. This will allow direct comparisons to be made with
other approaches, such as simultaneous analysis of concatenated
data matrices and compartmentalization methods (references above;
also [137]. There is a full spectrum of approaches for concatenating
analyses at different scales:
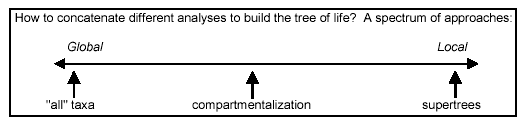
At the left end of this spectrum, the approach is to include all
possible OTUs and potential characters in one matrix. Generally
this is not actually done, because the sheer amount of data (millions
of possible OTUs) makes thorough phylogenetic analysis computationally
impossible. The most common approach is to select a few representatives
of a large, clearly monophyletic group (the exemplar method). Care
is sometimes taken to select representatives that are "basal"
OTUs within the group to be represented; however, this still does
not avoid two important problems: (i) within-group variation is
not fully represented in the analysis, and (ii) an increase both
in terminal branch lengths and in asymmetry between lengths of different
branches is introduced. These problems can lead to erroneous branch
attractions in global analyses.
At the right end of the spectrum, local analyses are simply grafted
together at the place where shared taxa occur, without reference
back to the original data. There are many ways to do this in detail
(as reviewed by Sanderson), but the important thing is that the
analyses on real character data are only done locally, and the concatenation
is based on the combination of local topologies rather than a combination
of local data sets into a global data set.
We will explore both of these approaches even though both seem
too extreme, one too global, the other too local. Thus we will also
explore a promising synthetic approach called compartmentalization
(by analogy to a water-tight compartment on a ship -- homoplasy
is not allowed in or out) that represents diverse yet clearly monophyletic
clades by their inferred ancestral states in larger-scale cladistic
analyses. A well-supported local topology is sought first, then
an inferred "archetype" or hypothetical ancestor (HTU)
for the group is inserted into a more inclusive analysis. In more
detail, the procedure we will use is to: (1) perform global analyses,
determine the best supported clades (these become the compartments);
(2) perform local analyses within compartments, including more taxa
and characters (more characters can be used within compartments
due to improved homology assessments among closely related organims);
(3) return to a global analyses, in one of two ways, either (a)
with compartments represented by single HTUs (the archetypes), or
(b) with compartments constrained to the topology found in local
analyses (for smaller data sets, this approach is better because
it allows character optimizations within each compartment).
The compartmentalization approach differs from the exemplar approach
in that the representative character-states coded for the archetype
are based on all the taxa in the compartment, thus the reconstructed
HTU is likely to be quite different from any real OTU. As an estimate
of the states of the most recent common ancestor of all the local
OTUs, the HTU is likely to have a much shorter terminal branch with
respect to the global analysis, which in turn can have the beneficial
global effect of reducing long-branch attraction. In addition to
these advantages of compartmentalization at the global level, the
local analyses will be better because one can: (1) include all local
OTUs for which data are available; (2) incorporate more (and better
justified) characters, by adding in those characters for which homology
could not be determined (aligned) globally; (3) avoid spurious homoplasy
that can change the local topology due to long-branch attractions
with distant outgroups. The effects of compartmentalization are
thus to cut large data sets down to manageable size, suppress the
impact of spurious homoplasy, and allow the use of more information
in analyses. This approach is self-reinforcing; as better understanding
of phylogeny is gained, the support for compartments will be improved,
leading in turn to refined understanding of appropriate characters
and OTUs.
Phylogenetic database enhancements.
As our data sets develop, we plan to assist larger efforts to develop
a new generation of phylogenetic data-bases, including TreeBase
and a pending ITR proposal for a national resource in phyloinformatics
(see management plan). The next generation of data resources needs
to be much more flexible than existing data bases (e.g., GenBank,
which is essentially "flat" with respect to phylogeny),
and sensitive to scale and the fractal nature of phylogenies (with
their many hierachically nested scales).
The exploration of the basic nature of phylogenetic data described
above will be applied to data-base research through modeling studies.
We will address fundamental questions about the nature of data before,
during, and after phylogenetic analysis. Biologists in this project
will work with collaborating computer scientists to model: (1) How
are elements of the data matrix (OTUs, characters, and states) defined
and recognized in any particular study? (2) How can heterogeneous
data types (e.g., DNA sequences, genomic rearrangements, morphology)
be compared/combined? (3) How can data sets and analyses at very
different scales be concatenated (e.g. supertree, compartmentalization,
or global approaches as discussed above)? (4) How can data sets
at these different concatenated scales, where OTUs are nested inside
larger ones and character definitions (e.g., alignments) change
as you move up and down the scale, be presented to the user community?
[previous] [next]
|



